Mass Spectrometry (MS)-Based Proteomics
Mass spectrometry-based proteomics is a powerful analytical technique that enables comprehensive identification, quantification, and characterization of proteins in complex biological samples. The technology operates by ionizing protein or peptide molecules and measuring their mass-to-charge (m/z) ratios, providing precise molecular weight information that can be used to determine protein composition, post-translational modifications (PTMs), and protein-protein interactions. The typical workflow involves sample preparation (protein extraction, digestion into peptides), chromatographic separation (usually via liquid chromatography, LC), ionization (electrospray ionization, ESI, or matrix-assisted laser desorption/ionization, MALDI), mass analysis (using high-resolution mass spectrometers such as Orbitrap or time-of-flight, TOF), and data processing with bioinformatics tools (e.g., MaxQuant, Proteome Discoverer). MS-based proteomics can be applied in discovery (shotgun proteomics) or targeted (selected reaction monitoring, SRM/parallel reaction monitoring, PRM) modes, allowing both unbiased protein profiling and precise quantification of specific targets. By comparing proteomic profiles between diseased and healthy states or before and after drug treatment, researchers can uncover dysregulated pathways, novel biomarkers, and potential therapeutic targets with high specificity and sensitivity.
Mass Spectrometry-Based Proteomics in Drug Target Discovery
Mass spectrometry-based proteomics has become indispensable in drug target discovery by revealing disease-associated protein networks and mechanisms of drug action. Zhang et al. (2016) demonstrated its power in NSCLC by uncovering AXL-mediated resistance to EGFR inhibitors through comprehensive phosphoproteomics.1 Similarly, Bai et al. (2020) mapped the Alzheimer's brain proteome, identifying GSK3β and synaptic proteins as promising intervention points.2 These examples demonstrate how proteomics bridges the gap between genomic findings and functional biology by directly measuring protein-level changes, validating drug targets, and identifying biomarkers for patient stratification. Furthermore, proteomics enables the study of PTMs (e.g., phosphorylation, ubiquitination) that are critical for cellular signaling and drug response, offering insights into combination therapies and resistance mechanisms.
Standard Mass Spectrometry-Based Proteomics Workflow
The application of MS-based proteomics in drug target discovery follows a structured pipeline:
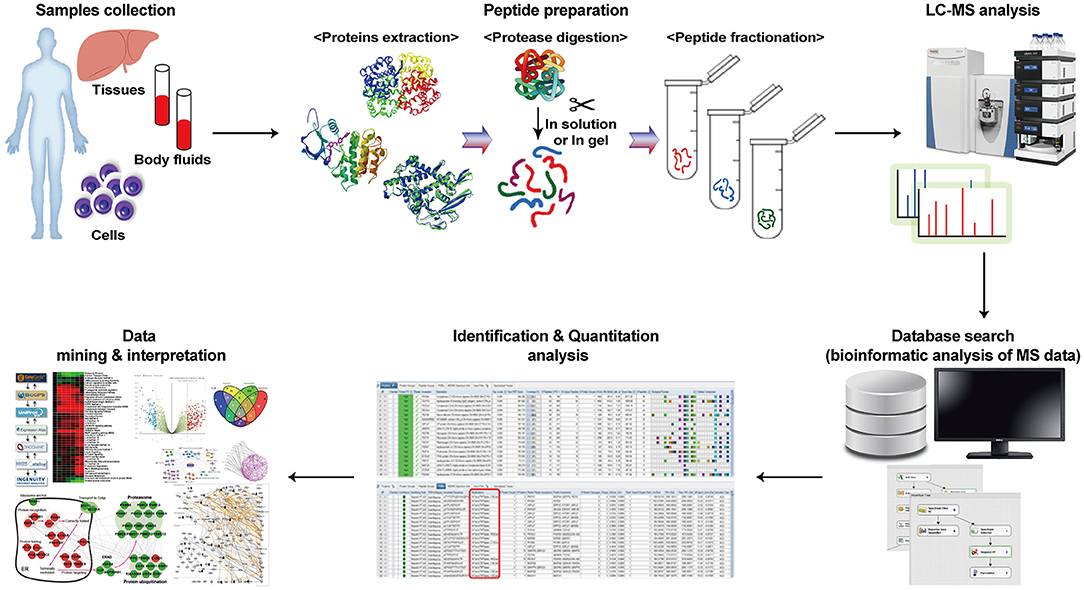 Fig 1. Workflow of the MS-based proteomics investigation.3
Fig 1. Workflow of the MS-based proteomics investigation.3
(1) Sample Preparation—extracting proteins from cells, tissues, or biofluids, followed by digestion (typically with trypsin) into peptides;
(2) Fractionation—using LC or gel-based methods to reduce sample complexity;
(3) Mass Spectrometry Analysis—acquiring high-resolution spectra via data-dependent acquisition (DDA) or data-independent acquisition (DIA) modes;
(4) Data Processing—identifying and quantifying peptides/proteins using database search algorithms (e.g., Andromeda, Sequest) and statistical tools (e.g., Perseus, limma);
(5) Bioinformatics & Pathway Analysis—annotating proteins with Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), or STRING networks to pinpoint dysregulated pathways;
(6) Target Validation—confirming candidates using orthogonal methods such as Western blot, siRNA knockdown, or cellular assays. This integrated approach ensures robust identification of high-confidence targets and accelerates translational research.
PharmaAnalytica's Technology Platform
Nano-1000 Micro-Spectrophotometer

The Nano-1000 microspectrophotometer ensures accurate protein/peptide concentration quantification (190-1100 nm) prior to LC-MS/MS analysis, enabling optimal sample loading and reproducible proteomics results.
LC-QTOF 7000

The LC-QTOF 7000 system delivers high-resolution, accurate-mass (HRAM) analysis for comprehensive protein identification and quantification, enabling deep proteome coverage and post-translational modification characterization in discovery proteomics. .
EXPEC 5800 LC-QTOF

EXPEC 5800 LC-QTOF combines quadrupole-time-of-flight (QTOF) technology with ultra-high mass resolution (<1 ppm) and wide dynamic range, enabling deep proteome profiling, PTM analysis, and precise quantification in MS-based proteomics.
PharmaAnalytica's MS-Based Proteomics Services
PharmaAnalytica offers state-of-the-art MS-based proteomics services designed to accelerate drug discovery with unparalleled depth, accuracy, and scalability. Our platform delivers:
Customized Solutions
Tailored workflows for membrane proteins, low-abundance targets, or post-translational modifications.
Quantitative Precision
Isobaric labeling or label-free quantification for reliable differential expression analysis.
Expert Bioinformatics
Advanced pipelines for statistical analysis, machine learning-based biomarker discovery, and druggability prediction.
Regulatory Support
GLP-compliant protocols for preclinical and clinical studies.
By leveraging PharmaAnalytica's expertise, pharmaceutical and biotech partners gain a competitive edge in target identification, mechanism-of-action studies, and biomarker development, ultimately shortening the path from discovery to clinical success.
References
- Chabon, J. J. , et al. (2016). "Circulating tumour dna profiling reveals heterogeneity of egfr inhibitor resistance mechanisms in lung cancer patients." Nature Communications. 7, 11815.
- Bai, B. , et al. (2020). "Deep multilayer brain proteomics identifies molecular networks in alzheimer's disease progression." Neuron. 105(6), 975.
- Kwon, Y. W. , et al. (2021). "Application of proteomics in cancer: recent trends and approaches for biomarkers discovery." Frontiers in medicine. 8, 747333.
Online Inquiry
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
Need More Information or Request A Quotation?
Related Links